INTRODUCCIÓN
El virus de Newcastle (NDV, de sus siglas en inglés Newcastle disease virus) es el agente causal de la enfermedad de Newcastle (ND), enfermedad altamente contagiosa con un amplio rango de huéspedes y reportada en todos los órdenes de aves (Seal et al., 1995). ND está distribuida a nivel mundial, donde es causante de notables pérdidas económicas para el sector avícola, y en el Perú está presente como enfermedad de reporte obligatorio (OIE, 2017). El NDV es un miembro de la familia Paramixoviridae, un virus envuelto que tiene un genoma de una sola hebra de ARN lineal de polaridad negativa. El genoma de ARN tiene un tamaño aproximado de 15 200 pb y contiene seis genes estructurales que en orden 3´ a 5´ del ARN genómico codifican, la proteína de nucleocápside (NP), fosfoproteína (P), proteína matriz (M), proteína fusión (F), hemaglutinina-neuramidasa (HN) y ARN polimerasa ARN dependiente (L) (Cattoli et al., 2011; Alexander et al., 2012).
NDV puede ser clasificado en cepas altamente patógenicas (velógenico), intermedias (mesogénica) y no patogénicas (lentogénicos) basados en su patogenia en pollos (Cattoli et al., 2011). La mejor estrategia de prevención es la vacunación con cepas atenuadas, la cual ha sido ampliamente usada por más de 50 años (Gallili y BenNathan, 1998). Actualmente, la producción de vacunas de NDV es principalmente realizada en sistemas de huevos embrionados libres de patógenos específicos (SPF) debido a su accesibilidad y bajo costo (Seene et al., 2003); por otra parte, la medición del título exacto es importante en la producción de vacunas virales, valiéndose de ensayos de hemaglutinación, ensayos en placa y de dosis media infectiva en tejidos (TCDI50) y embriones (DIEP50). Sin embargo, estos métodos pueden no ser altamente específicos y consumir un tiempo considerable (McGinnes et al., 2006).
La técnica de qPCR tiene muchas ventajas sobre los métodos tradicionales, incluyendo rapidez, posibilidad de cuantificación, alta sensibilidad y especificidad (Jang et al., 2011). Varios métodos de RT-qPCR han sido desarrollados para detectar y cuantificar NDV (Seal et al., 1995; Tan et al., 2009; Fuller et al., 2010; Jang et al., 2011). Sin embargo, la mayoría de estos métodos están enfocados en el diagnóstico de aves infectadas con ND, pero no todos incluyen criterios de optimización específicos. Por tanto, se requiere optimizar una RT-PCR que pueda ser utilizada para cuantificar el NDV utilizado en la producción de vacunas aviares.
Este estudio tuvo por objetivo desarrollar una plataforma molecular para la cuantificación del NDV a partir de un sistema de cultivo en huevos embrionados SPF. Para lograr esto, se amplificaron, a través de RT-PCRc, diferentes regiones del genoma viral utilizando cebadores dirigidos hacia los genes F, NP, L y M del NDV. Los cebadores que cumplieron con las características/criterio de selección (Innis y Gelfand, 1999; Ottiger, 2010) fueron utilizados para cuantificar el virus a través del RT-qPCR.
MATERIALES Y MÉTODOS
Cepas Virales
La cepa vacunal NDV LaSota forma parte del cepario de los Laboratorios FARVET (Perú), la cual fue reactivada en huevos embrionados de pollo SPF de 9 días de edad por ruta alantoidea. Luego el fluido alantoideo fue centrifugado a 3000 g por 15 min y el sobrenadante fue almacenado en alícuotas a -80 °C hasta su uso. El ARN genómico del virus de la bronquitis infecciosa aviar (IBV), Metapneumovirus aviar (SHS) y el ADN genómico del virus de la laringotraqueitis infecciosa (ILTV), y de las bacterias Avibacterium paragallinarum, Gallibacterium anatis y Ornithobacterium rhinotracheale forman parte del cepario de los Laboratorios FARVET SAC.
Cebadores
Se seleccionaron cebadores candidatos dirigidos a las regiones F, NP, L y M del virus NDV LaSota, en base a revisión bibliográfica (Seal et al., 1995; Farkas et al., 2009; Fuller et al., 2010; Cattoli et al., 2011). El criterio de inclusión fue la capacidad de detectar, por lo menos cepas de NDV de Clase I. Los cebadores fueron sintetizados de forma comercial (IDT, EEUU). Las secuencias de los cebadores y otros detalles están descritas en el Cuadro 1. Los cebadores fueron denominados siguiendo la abreviatura del virus, el gen al cual fue dirigido y un número. Se comprobó su ubicación en la cepa vacunal utilizada, empleando el programa SnapGene Viewer v. 3.3.3 (GSL Biotech, EEUU) (Figura 1).
Producción y Purificación de NDV
Se infectaron huevos embrionados SPF de 9 días con la cepa NDV LaSota a una multiplicidad de infección (MOI) de 0.01 e incubados a 37 °C durante 96 horas. El sobrenadante fue cosechado y centrifugado a 3000 g durante 30 min a 4 °C para eliminar el debris celular. Luego, fue pasado por un filtro de 0.22 μm. El título de NDV del fluido alantoideo fue determinado por hemaglutinación (McGinnes et al., 2006; OIE, 2017) y usado como inóculo para posteriores experimentos.
Para la purificación del virus, el fluido alantoideo de NDV previamente clarificado fue ultracentrifugado en botellas de 250 ml en el rotor Type 19 de la ultracentrífuga Optima L-100K (Beckman Coulter, EEUU) a 48 000 g durante 18 horas. El sobrenadante fue descartado y los pellets fueron resuspendidos en 5 ml de PBS 1X. Luego, fueron transferidos a tubos de policarbonato previamente llenados con sucrosa al 30%(P/V) (Sigma Aldrich, EEUU), centrifugados en el rotor SW28 a 11 000 g durante 18 horas. Se descartó el sobrenadante y el pellet viral fue usado para el aislamiento de ARN de NDV.
Cuadro 1. Cebadores evaluados para el desarrollo de la plataforma molecular para la cuantificación del virus de Newcastle

 Figura 1.
Figura 1. Ubicación de los cebadores candidatos en el genoma de la cepa vacunal NDV LaSota (Código de accesión GenBank N.° AF077761) con tamaño de genoma referencial de 15 186 pb
Aislamiento de ARN Genómico Viral
El pellet viral fue extraído con el RNeasy Midi Kit (Qiagen, Alemania), de acuerdo con las instrucciones del fabricante. Los productos obtenidos se conservaron a -80 °C hasta su uso.
Preparación de Material Genómico de NDV LaSota
El ARN genómico de cadena sencilla (ssARN) de NDV LaSota fue sometido a diluciones seriadas desde 1 ng hasta 100 ag por cada μl de ARN genómico, procedimiento que también se siguió para el cADN obtenido por retrotranscripción reversa que fue llevada a cabo con el kit comercial ProtoScript® First Strand cDNA Synthesis Kit (New Englands Biolabs, UK) en el termociclador MasterCycler Pro-S (Eppendorf, Alemania). El cADN obtenido se cuantificó por fluorometría en el Qubit (Invitrogen, EEUU). La pureza fue determinada por la relación A260/A280 en el Biophotometer plus (Eppendorf, Alemania) para ambos tipos de ácidos nucleicos.
La cuantificación de NDV presente en el fluido alantoideo fue llevada a cabo en el Qubit 1.0 Fluorometer (Invitrogen, EEUU) y para determinar los equivalentes en copias virales teóricas se usó la siguiente fórmula (Ke et al., 2006): Y = (moléculas/μl)=X (g/l) (tamaño del genoma x 340) x 6.02 x 1023 para el material de ARN y la formula Y = (moléculas/μl)=X (g/l) (tamaño del genoma x 330) x 6.02 x 1023 para material de cDNA. Con base a estos valores se construyó una tabla de equivalencias (Cuadro 2).
Optimización de la RT-PCR Convencional
El RT-PCRc fue desarrollado en dos pasos: la transcripción reversa para la obtención cADN fue llevada a cabo con el Kit ProtoScript (New England Biolabs, EEUU) de acuerdo con las instrucciones del fabricante.
Brevemente, en un volumen final de 20 μl usando 5 μl de ARN total, 2 μl de Ramdom hexamer primer (600 μM), 2 μl de ditiotreitol (100 μM), inhibidor de ARNasa murina (40 u/μl) y 200 U de MMLV (transcriptasa reversa del virus de la leucemia Murina de Moloney). La retrotranscripción se llevó acabo en un termociclador Mastercycler®Pro-S (Eppendorf, EEUU) a 25 °C durante 5 min y posteriormente a 42 °C durante 3 horas. Luego se cuantificó por fluorometría (Qubit 1.0 Invitrogen, EEUU) y se ajustó la concentración de cDNA deseada tomando al final 2 μl para la PCR que se realizó en el termociclador mencionado. Se empleó el kit GoTaq Flexi DNA-Polimerasa (Promega, EEUU), con deoxyribonucleósido trifosfato (dNTPs) a una concentración de 200 μM, MgCl2 2.0 mM, GoTaq DNA Polimerasa (1.25 u) y GoTaq Flexi Buffer 1x.
La denaturación inicial a 94 °C x 5 min, 35 ciclos de 90 °C x 30 s, (45-65 °C) x 30 s, 72 °C x 30 s y un paso de extensión final a 72 °C por 5 min. Se optimizaron las condiciones de PCR, donde se evaluó temperatura de hibridación y sensibilidad para la totalidad de los cebadores candidatos y, luego, al par de cebadores con las condiciones ideales se ajustó la concentración de MgCl2 y de los cebadores (Innis y Gelfand, 1999). El cADN de fluido alantoideo de huevos SPF fue utilizado como control negativo y el agua de grado PCR fue utilizada como blanco, los cuales fueron incluidos en cada corrida.
Los productos de PCR fueron corridos en un sistema de electroforesis horizontal Compact M (Analityk Jena, Alemania). Los geles fueron preparados al 1.5% con agarosa (Cleaver Scientific, UK), buffer TAE y SyberSafe (Invitrogen, EEUU) electroforetizados a 80 mV durante 75-90 min y visualizados en un transiluminador de luz azul Safe Imager 2.0 (Invitrogen, EEUU). Se emplearon los marcadores de peso molecular 100 bp y O’Gene Ruler (Thermo Scientific, EEUU).
Cuadro 2. Equivalentes en copias virales teóricas del cADN/μl obtenido por transcripción reversa y el ssARN procedente de la purificación de los ácidos nucleicos de NDV. Se tomó como genoma de referencia la cepa NDV LaSota (Código de accesión GenBank N° AF077761) con tamaño de genoma referencial de 15 186 pb

Optimización de la RT-qPCR en un Paso Usando Sybr Green I
En base al par de cebadores del gen candidato que cumplió con los parámetros optimizados de temperatura de hibridación, sensibilidad, concentración de cebadores, concentración de MgCl2 y especificidad, se desarrolló el RT-qPCR utilizando el kit KAPA SYBR FAST One-step for LightCycler 480 Mix (Kapa Biosystems, EEUU), donde cada reacción de PCR fue llevada a cabo tomando ARN genómico purificado. El protocolo de RT-qPCR consistió en 40 ciclos de 95 °C durante 10 s, hibridación de 60 °C por 20 s y extensión de 72 °C por 20 s. Las curvas de disociación consistieron en 95 °C por 5 s, 65 °C por 30 s y calentado hasta 97 °C a una velocidad de 0.1 °C/s. Los productos de amplificación fueron detectados mediante análisis de curva de disociación y confirmados por secuenciación (Macrogen, Corea). Los valores del ciclo de amplificación (Cp) y temperatura de disociación (Tm) fueron analizados con el Software LightCycler 480 v. 1.5.0. Los análisis estadísticos fueron realizados con el programa Microsoft Excel v. 2013.
Ensayos de Sensibilidad
Para los ensayos de sensibilidad se realizaron diluciones decimales seriadas de cADN/ssARN para la RT-PCRc y RT-qPCR, respectivamente, que fueron desde 10 ng hasta 1 ag/μl. Los ensayos se realizaron por triplicado durante tres días consecutivos. Para la RT-PCRc se empleó el termociclador MasterCycler (Eppendorf, Alemania), y para la RT-qPCR se usó el temociclador Light Cycler 480 (Roche, EEUU). Cada dilución fue corrida hasta 10 veces durante 3 días y se determinó la sensibilidad como la dilución previa a aquella que poseía un coeficiente de variación > 5%. Se tomó en cuenta la presencia de banda de interés o la formación de curvas de fluorescencia con valor Ct < 35 y/o la presencia de pico de disociación para el RT-PCRc y RT-qPCR, respectivamente.
Ensayos de Especificidad
Para estimar la especificidad del método se emplearon varios genomas virales y bacterianos patológicamente relacionados: IBV, SHS, ILTV, Avibacterium paragallinarum, Ornithobacterium rhinotracheale y Gallibacterium anatis. Se empleó un fragmento amplificado para NDV LaSota en el RT-PCR y un pico específico de disociación para el RT-qPCR (Jennings et al., 2009; Berwouts et al., 2010; Mattocks et al., 2010). Los ensayos se realizaron por triplicado. 0
El flujograma de actividades implementadas en el estudio se muestra en la Figura 2.
Figura 2. Flujograma de trabajo para el desarrollo del estudio
RESULTADOS
Una de las primeras fases para el proceso de estandarización de métodos moleculares (Figura 2) de RT-PCR fue determinar la temperatura ideal en la cual los cebadores hibridan con la secuencia de nucleótidos target, para lo cual se realizó una gradiente de temperaturas. En la Figura 1 se observan los resultados obtenidos por ensayos empíricos para los cuatro cebadores candidatos, donde los cebadores NDV 1F y NDV 5M presentan un amplio rango de temperaturas (45.7 a 61.6 °C y 51.3 a 63.4 °C, respectivamente), con la banda de interés definida y sin presencia de productos inespecíficos de alto o bajo peso molecular. El hecho de que puedan hibridar a temperaturas elevadas disminuye la presencia de bandas inespecíficas, mientras que los cebadores NDV 3NP y NDV4L presentaron un estrecho rango de temperaturas a elegir (47 a 59.2 °C y 45.7 a 56.6 °C, respectivamente), donde las temperaturas más bajas podrían propiciar la formación de inespecificidades, pese a que las bandas del producto de interés fueron definidas (Figura 3). Las temperaturas de hibridación optimizadas fueron 59.2, 56.6, 53.9 y 61.6 °C para los cebadores NDV 1F, NDV 3 NP, NDV 4L y NDV 5M, respectivamente.
En los ensayos de sensibilidad se determinó la cantidad del genoma de NDV, en su equivalente de cADN, que podía ser detectado con cada par de cebadores. En la Figura 4 se aprecia que los cebadores NDV1F, NDV5M y NDV4L son capaces de detectar hasta 10 fg de cADN, mientras que los cebadores NDV 3NP son 10 veces menos sensibles. Si bien es cierto que tres pares de cebadores tuvieron el mismo nivel de sensibilidad,
los cebadores NDV 5M pudieron hibridar a una temperatura superior a 60 °C, lo que mejoraría la discriminación del fragmento de interés y reduciría la formación de dímeros entre cebadores, por lo que se consideraron para la optimización final.
Para la estandarización del RT-PCRc en dos pasos se optimizó la concentración de MgCl2 y de los cebadores NDV 5M. Con respecto al MgCl2 se puede notar que la concentración ideal fue de 2.5 mM (Figura 5A), mientras que para la concentración de cebadores fue 0.25 μM (Figura 5B).
Con las concentraciones de los componentes de la reacción de RT-PCR convencional optimizada se llevó a cabo un segundo ensayo de sensibilidad en donde se pueden apreciar el producto de PCR de interés desde 10 ng hasta 1 fg, sin amplificados inespecíficos de alto o bajo peso molecular (Figura 6), por lo que el protocolo final tuvo las siguientes condiciones: denaturación inicial a 94 °C por 5 min, 35 ciclos de 90 °C por 30 s, 45-65 °C por 30 s, 72 °C por 30 s y un paso de extensión final a 72 °C por 5 min.
Figura 3. Ensayos de gradiente de temperaturas para los cebadores candidatos: A: NDV 1F; B: NDV5 M; C: NDV 3NP; D: NDV 4L
 Figura 4.
Figura 4. Ensayos de sensibilidad para los cebadores candidatos. Diluciones decimales desde 10 ng hasta 10 ag: A: NDV 1F; B: NDV 5M; C: NDV 3NP; D: NDV 4L
 Figura 5.
Figura 5. Estandarización del RT-PCRc para los cebadores NDV 5 M: A. Optimización de las concentraciones de MgCl2. B. Optimización de la concentración de cebadores
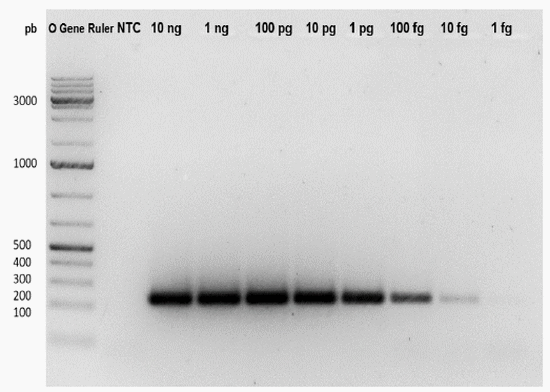
Figura 6. Visualización de los productos de RT-PCRc para los cebadores NDV 5 M. Se usó el marcador de peso molecular O’Gene Ruler
La especificidad depende de la secuencia de los cebadores. Por tal motivo, en los ensayos de especificidad para el RT-PCRc se observó que no hubo amplificación para el material genómico de otros virus y bacterias respiratorias, demostrando que los cebadores NVD 5M son adecuados por no brindar reacciones cruzadas frente a material genómico de otros patógenos aviares (Figura 7).
Luego de optimizar la plataforma de detección por RT-PCRc en dos pasos, se llevó a cabo los ensayos de RT-qPCR en un paso, cuyo programa consistió en 5 min de retrotranscripción reversa, 3 min para la activación de la enzima hot-start y 40 ciclos de 95 °C por 10 s, hibridación de 60 °C por 20 s y extensión de 72 °C por 20 s. Se observa que el RT-qPCR tuvo una sensibilidad de hasta 1 fg/μl, que pudo ser discriminada en base a la curva de disociación (Figura 8). Esto demuestra que la técnica de RT-qPCR es 10 veces más sensible que el método convencional; sin embargo, al introducir material genético del huésped se reduce la sensibilidad hasta 10 veces (Figura 9).
Los ensayos de especificidad para el RTqPCR demostraron presencia de un pico de interés con una temperatura mayor a 82 °C a diferencia de los picos generados por otras cepas patógenas respiratorias (72-80 °C) (Figura 10). El uso de agentes fluorescente como el Sybr Green I exige el uso de estas curvas para la confirmación de los verdaderos positivos. Esta técnica es sencilla de realizar y menos costosa en comparación con las sondas fluorescentes.
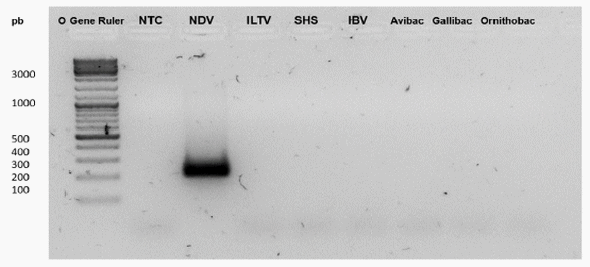
Figura 7. Ensayos de especificidad del RTPCRc utilizando los cebadores NDV 5M para patógenos respiratorios virales y bacterianos: No Template control (NTC), NDV, ILTV, SHS, IBV, Avibacterium paragallinarum, Gallibacterium anatis,Ornithobacterium tracheale. Se usó el marcador de peso molecular O’Gene Ruler. Todas las cepas, estuvieron normalizadas a una concentración de 1 pg/μl
DISCUSIÓN
En este estudio se ha desarrollado una plataforma específica y sensible de RT-qPCR para la detección y cuantificación de NDV producido en un sistema de huevos embrionados SPF. La búsqueda bibliográfica dio como resultado la presencia de al menos cuatro pares de cebadores para detectar NDV de clase I (Farkas et al., 2009; Fuller et al., 2010). Estos cebadores fueron sometidos a una evaluación para determinar el más apropiado para la detección de NDV en muestras de fluido alantoideo. Se comparó la temperatura de hibridación óptima sugerida por el fabricante con la temperatura óptima determinada por análisis empírico (Rochelle et al., 1997), indicando los resultados que la temperatura optimizada fue mayor (56.6 y 61.6 °C) para los cebadores NDV 3NP y NDV 5M y menor (59.2 y 53.9 °C) en los cebadores NDV 1F y NDV 4L, difiriendo con la calculada por el fabricante. Resultados similares fueron encontrados por Rochelle et al. (1997) al evaluar seis pares de cebadores para la detección de Cryptosporidium parvum y Giardia lamblia.
Los cebadores NDV 5M obtuvieron los mejores resultados, debido a que las temperaturas de hibridación elevadas mejoran la discriminación entre dímeros de cebadores y extensión con nucleótidos erróneos en el extremo 3´ (Figura 3B). Esta característica y su capacidad para detectar hasta 10 fg de cDNA de NDV le permitieron continuar las siguientes etapas de optimización que incluyeron la gradiente de concentración de MgCl2 y cebadores, determinándose que la concentración de ambos componentes se encontraba dentro de los rangos establecidos (Innis y Gelfand, 1999).
Para incluir a los cebadores NDV5M en el desarrollo de la plataforma de RT-qPCR en un paso, se optimizó en el protocolo de PCR el tiempo de extensión, de 1 a 20 s, obteniendo mejores resultados con respecto a lo indicado por el fabricante (datos no mostrados). Luego se determinaron la sensibilidad y especificidad. Acorde a la literatura, los cebadores elegidos NDV 5M han demostrado ser uno de los más conservados (Cattoli et al., 2011) en comparación con las otras secuencias. Los cebadores NDV3P (Tan et al., 2004) fueron diseñados para la proteína F, obteniendo una baja sensibilidad (desde 10 ng hasta 10 pg/μl). Tan et al. (2009) utiliza ron los NDV3P, asumiendo que la proteína NP es más conservada que la proteína F; sin embargo, su ensayo no mostró alta sensibilidad (109 a 105 copias de DNA plasmídico/ reacción) por lo que no formaron parte del protocolo final. Los cebadores NDV 4L, diseñados por Fuller et al. (2010) tuvieron alta sensibilidad, pero mostraron inespecificidades.
El uso de ssARN genómico purificado en lugar de plásmidos reflejan las condiciones naturales en las cuales es hallado el virus, lo cual es una ventaja en la determinación de la sensibilidad. Por otro lado, los cebadores de NDV 1F diseñados por Jang et al. (2011) presentaron bandas de interés menos definidas y su sensibilidad solo fue de aproximadamente de 400 copias virales/μl. Si bien es cierto, estos autores desarrollaron un protocolo de RT-qPCR para la detección y cuantificación de la cepa vacunal NDV LaSota producida en un sistema de biorreactores, se ha demostrado que este tipo de sistemas genera, por lo general, menores títulos virales que los alcanzados en huevos SPF, además de ser económicamente menos accesible (MacGuines et al., 2006). A su vez, los cebadores NDV5M diseñados por Seal et al. (1995), orientados hacia la proteína matriz, tuvieron como objetivo caracterizar cepas de NDV de campo por secuenciación directa y filogenia por lo cual su sensibilidad no fue determinada. En este estudio se determinó su sensibilidad para la cepa vacunal NDV LaSota, llegando a detectar hasta 116 copia virales/μl (1 fg/μl) por RTqPCR, demostrando que el gen M es una de los más conservados (Cattoli et al., 2011).
Los resultados de especificidad demostraron que no hubo amplificación para virus y bacterias patogénicamente relacionados. El desarrollo de esta plataforma de cuantificación por técnicas moleculares con condiciones optimizadas se puede emplear en el proceso de monitoreo de carga viral en la producción de vacunas de NDV LaSota.
 Figura 8.
Figura 8. RT-qPCR para los cebadores NDV 5M. A. Amplificación de las curvas de sensibilidad. B. Curvas de disociación (melting) de productos obtenidos con diferentes concentraciones de virus.
Figura 9. Curvas de disociación (melting) de productos obtenidos con diferentes concentraciones de NDV mediante RT-qPCR en tiempo real. A. Curvas sin presencia de material genético (ARN) del huésped. B. Curvas con presencia de material genético (ARN) del huésped
 Figura 10.
Figura 10. Especificidad del RT-qPCR en tiempo real. Análisis de la curva de disociación (melting) de productos de RT-qPCR en tiempo real. Solo la cepa de NDV muestra un pico de disociación de elevada temperatura (Tm > 82 ºC)
CONCLUSIONES
Se desarrolló una plataforma molecular en base al gen matriz (M) para la cuantificación NDV a partir de un sistema de cultivo en huevos embrionados SPF. Se optimizaron los parámetros del RT-PCRc y se continuó con el desarrollo de RT-qPCR, el cual fue 10 veces más sensible que el RTPCRc.














